Structural Bioinformatics Research
Predicting Protein-Protein Interactions
Almost all biological processes are mediated by protein-protein interactions (PPI). As a consequence, PPIs are often involved in disease mechanisms and deeper knowledge on such PPIs could be very helpful to study and develop therapeutic approaches. However, our current knowledge of PPIs is still lacking important pieces. One such piece is the limited availability of detailed structural information on protein complexes. Here, the structural bioinformatics team is involved in the development of methods to predict complex structures of PPIs using computational, experimental, as well as hybrid approaches. More precisely, the aim of these methods is to detect interacting residues, thus allowing to efficiently narrow down the interacting surface patches of the proteins. Penultimately, these informations can be used as distance constraints to guide standard protein-protein docking allowing to overcome several of its weaknesses.
Computational Approaches
In collaboration with the Marks Lab at Harvard Medical School, we have been co-developing EVcomplex (Hopf, T.; Schärfe, C. et al. 2014), a method to predict the interacting residues of PPIs from sequence alignments through identification of evolutionary couplings. In order to improve the existing methods we develop integrative template-free modelling and supervised learning approaches that try to fully leverage the sparse information on interacting residues stored in related sequences. Additionally, we try to further include various geometrical and physicochemical properties of known protein complexes to improve the quality of predicted residue pairs.
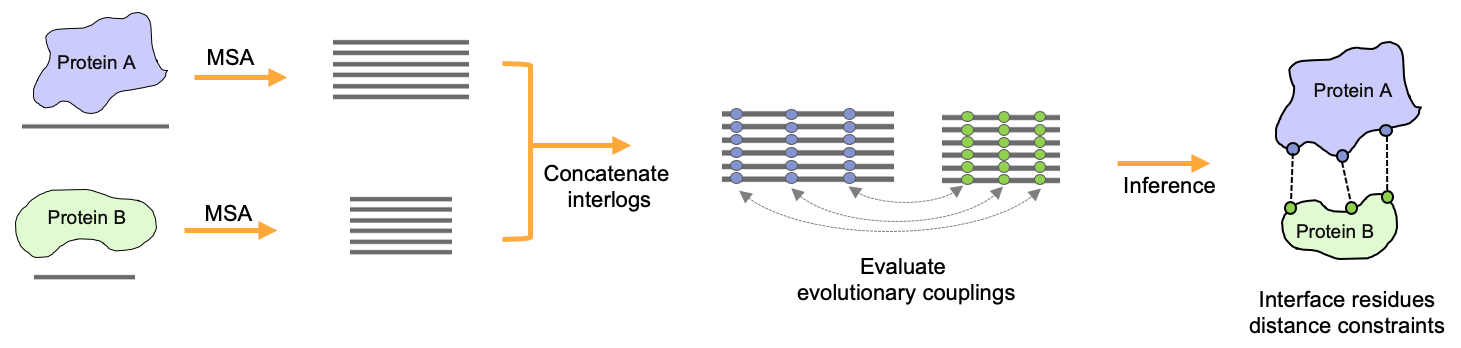
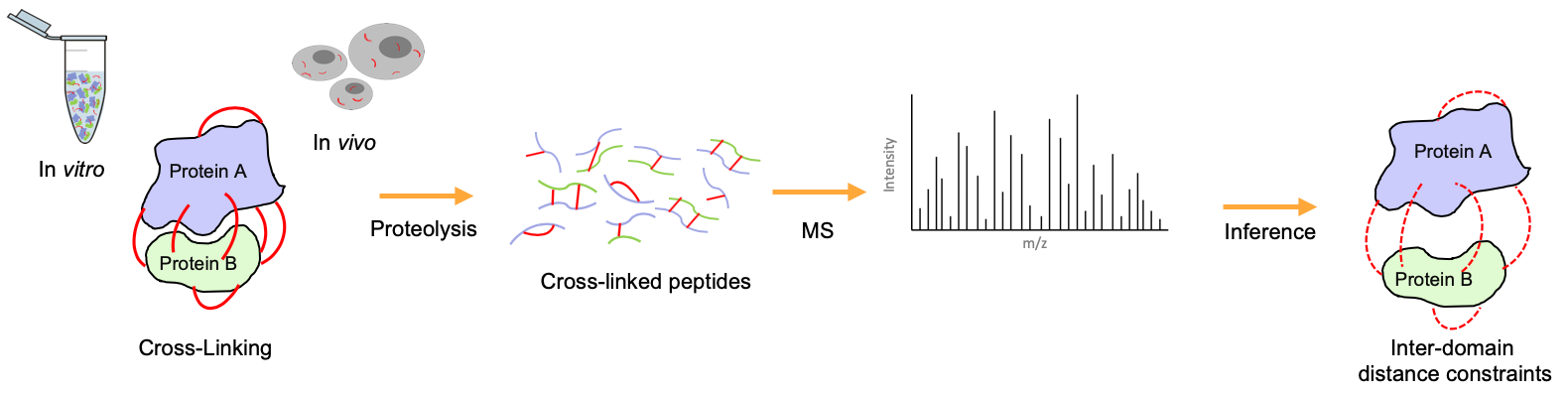
Experimental Approaches
Chemical cross-linking coupled
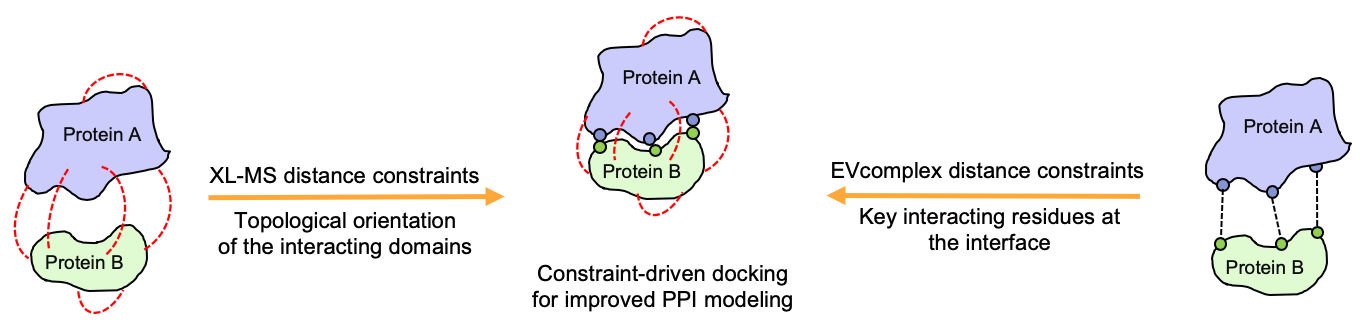
Hybrid Approaches
Having access to both, predicted and experimentally retrieved inter-protein residue distance constraints, we try to use both data sources in order to develop hybrid approaches for the prediction of protein complexes with enhanced quality when compared to the purely theoretical or experimental approaches.
Computer-Aided Drug Design
CADD is a scientific field where computer science meets structural biology, chemistry, the pharmaceutical sciences, as well as genomics. The aim is to understand how drugs work, to provide tools to assist the development of new drugs, and penultimately to develop methods that allow the prediction of new drug candidate molecules in silico.
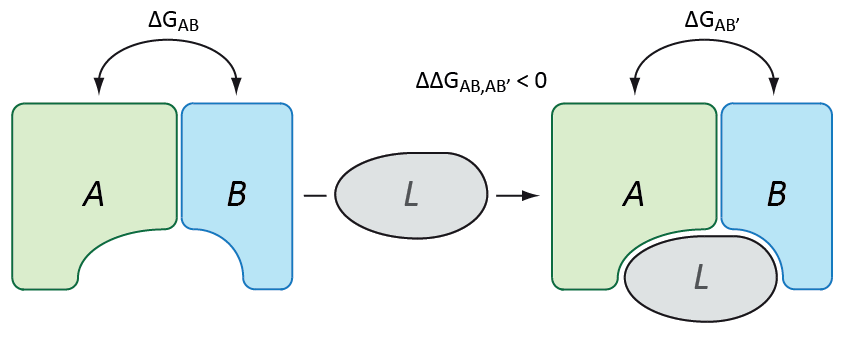
Stabilization of Protein-Protein Interactions
As already described above, PPIs are a fundamental mechanistic feature within biological systems. Thus, impaired PPIs can be the cause of various diseases. One possible disease scenario is that a required protein complex is not available in a sufficient amount, which can be due to several reasons. In order to increase the concentration of such a complex one can try to shift the equilibrium from unbound protein monomers to the bound state. This, however, effectively requires to increase the attraction of the binding partners to each other, i.e. binding free energy has to be decreased. This effect is indeed achieved through a fascinating mechanism of action of small molecules that are able to stabilize PPIs (Thiel, P. et al. 2012). In order to identify PPI-stabilizing candidate molecules we develop methods an protocols that allow to assess the stabilizing activity of small molecules for a given PPI and to enable stabilizer-tailored VS.
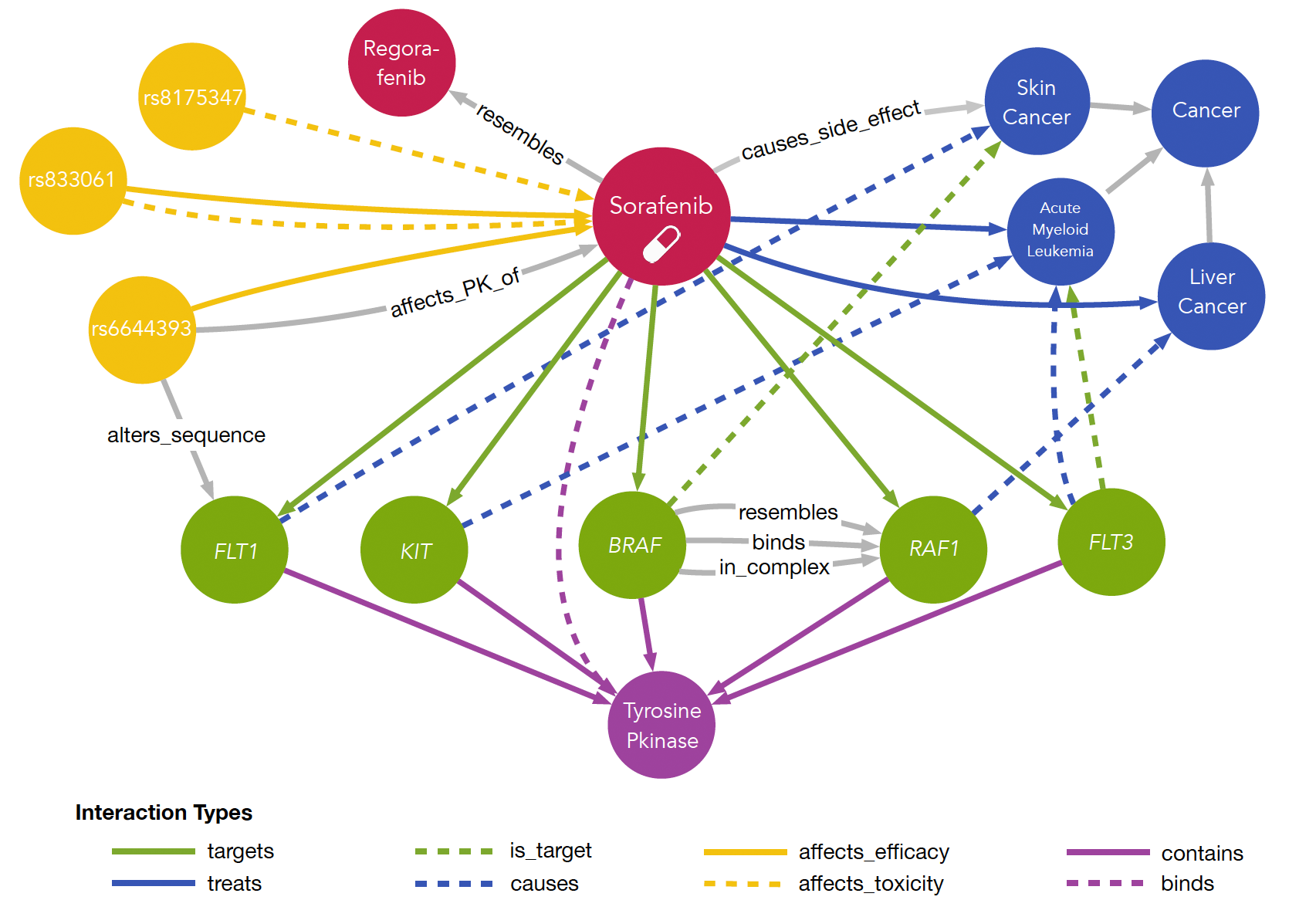
Pharmacogenomics
Genetic variation within the human population is an important factor that can highly affect the treatment effectiveness of drugs. Studying this genomic role for drug response is termed pharmacogenomics. In this context, we have been studying the extent to which drugs and drug-related genes are affected by genetic variation (Schärfe C. et al. 2017), focusing on known drug target and ADME genes. Including
Drug Repurposing
Trying to identify new indications for existing drugs or failed drug candidates is an interesting strategy to bypass major steps of the classical drug discovery pipeline. Due to lack of the discovery phase and the availability of rich pharmacokinetic and toxicity information this approach has the potential to significantly safe time and money. In order to perform drug repurposing one strategy is trying to collect all available information on diseases, drugs, their targets, associated PPIs and genes as well as their genetic variations. Integrating such data into a single information resource allows to study networks formed by these entities either locally by focusing on a selected entity or on a global scale. We have exactly been doing this and developed myDrug, an integrated platform to perform drug repurposing.
Software Development
In course of the work described

